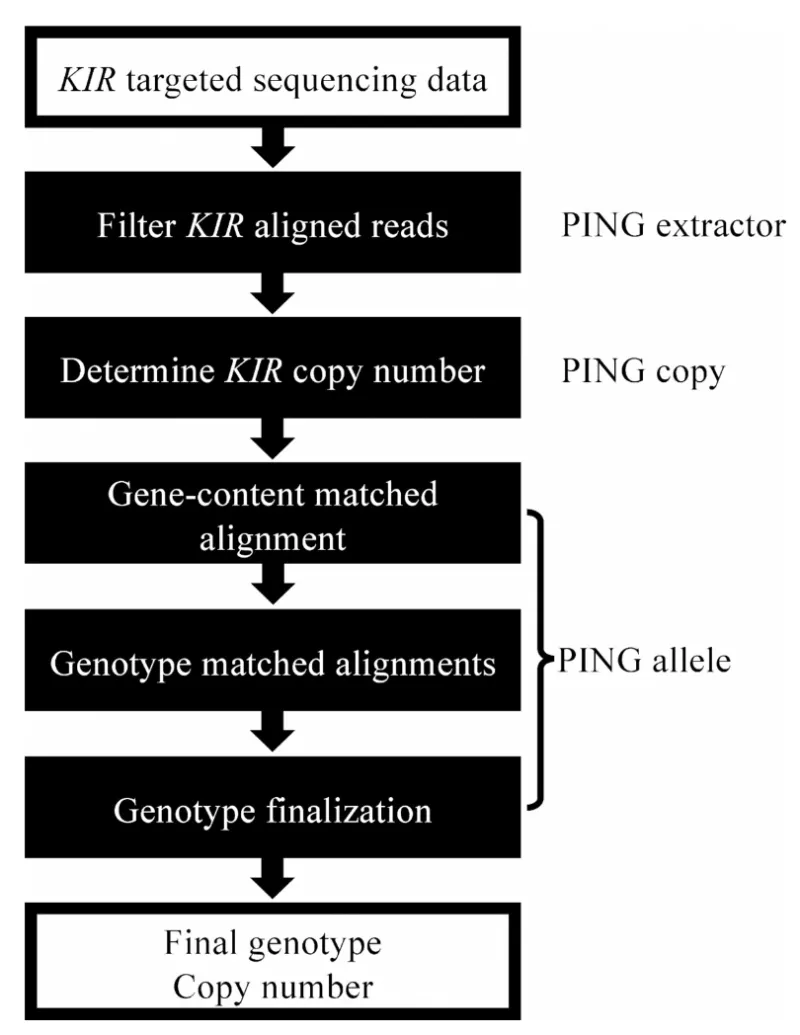
PING is free software used for genotyping the highly polymorphic killer immunoglobulin-like recptor (KIR) loci. The PING pipeline, developed in collaboration with Dr. Paul Norman (CU Anschutz), was created to automatically extract KIR reads from whole genome or exome data sets, attribute them to specific loci and generate a consensus sequence that can be interpreted as a KIR genotype. The PING analysis pipeline is designed to detect all known and any novel KIR SNP variants, a crucial limitation of all other methods. Using filters that consider all variants of each homologous gene, including pseudogenes, the method ensures selection of only those read-pairs mapping exclusively to a locus. All procedures are performed using free software and R language programming. More recently, an updated version of PING was published to provide KIR interpretation from whole genome sequence (WGS) data to increase the utility of WGS datasets, which has become a standard sequencing approach.
A full description of the pipeline can be found in Marin et al. 2021, PLOS CompBio and a full description of the updated version of this software can be found in Marin et al. 2023. PING was developed with the support of NIH grant R01AI128775.